

ALS
ALS
Omfattning av kunskapsstödet
Vårdförloppet inleds vid misstanke om amyotrofisk lateralskleros (ALS) hos en vuxen patient och avslutas när misstanken har avskrivits eller när patienten avlider.
En fullständig beskrivning av kriterierna för vårdförloppets omfattning finns under avsnitt Ingång och utgång.
Vårdnivå och samverkan
Misstanke om ALS kan väckas både inom primärvård och specialistvård. Så snart en sådan misstanke uppstår remitteras patienten omgående till neurolog för specialiserad utredning. Mer information finns under åtgärderna A – Initial utredning och C – Remiss till neurologimottagning.
En kontinuerlig samverkan mellan involverade vård- och omsorgsgivare är central och baseras på patientens aktuella behov. Exempel på aktörer är neurologimottagning och annan specialiserad vård, primärvård, palliativa team, assistansbolag, Försäkringskassan och omsorgsverksamheten, som vård- och omsorgsboende och hemtjänst.
Relaterade kunskapsstöd
Om hälsotillståndet
Definition
I detta vårdförlopp används termen amyotrofisk lateralskleros (ALS) som ett samlingsnamn för klassisk ALS och de närbesläktade formerna progressiv spinal muskelatrofi (PSMA) och primär lateralskleros (PLS). ALS, PSMA och PLS är exempel på motorneuronsjukdomar. Vid klassisk ALS påverkas både övre och nedre motorneuron, medan PSMA endast påverkar de nedre motorneuronen och PLS de övre motorneuronen.
ALS kännetecknas av progressiv och asymmetrisk muskelsvaghet. Sjukdomen debuterar i en arm eller ett ben hos cirka 65 % av patienterna. Hos knappt en tredjedel av patienterna börjar sjukdomen i bulbär region (mun och svalg) vilket leder till tal- och sväljningspåverkan. I ett fåtal fall debuterar ALS med andningspåverkan och ytterligare några med frontotemporal demens (1) (2).
Förekomst
Incidensen av ALS i Sverige har ökat över tid och uppskattas nu till cirka 4 fall per 100 000 personår (3), vilket motsvarar cirka 420-430 nydiagnostiserade patienter per år. Punktprevalensen i en studie baserad på data från Stockholm år 2013-2014 var 6,6 respektive 7,9 fall per 100 000 invånare (4). Om dessa värden antas gälla nationellt och vara oförändrade sedan dess motsvarar det cirka 700–840 patienter i riket. Det nationella kvalitetsregistret för ALS, ALS-registret, som är ett delregister i Svenska Neuroregister och ibland benämns MND-registret, anger en högre förekomst: omkring 970 patienter, baserat på en opublicerad inventering från år 2016 (832 patienter) samt en antagen årlig ökning med 2 % (5).
Orsaker
Hos cirka 97 % av personer med ALS förekommer inklusioner som innehåller transactive response DNA-binding protein 43 (TDP-43) i nervcellernas cytoplasma. Mekanismerna bakom denna aggregering är inte helt klarlagda, men faktorer som till exempel förändringar i proteinmetabolismen, mitokondriell dysfunktion, oxidativ stress och neuroinflammation misstänks bidra (1).
Riskfaktorer
ALS anses uppstå genom en kombination av genetiska faktorer och miljöfaktorer.
Cirka 10 % av alla personer med ALS rapporterar att sjukdomen finns i släkten. I dessa fall orsakas sjukdomen oftast av ett sjukdomsanlag med hög genomslagskraft. DNA-analyser visar att ytterligare cirka 15 % av personer med ALS är bärare av ett sjukdomsanlag med nedsatt genomslagskraft (6), recessiv nedärvning (7) eller har en de novo-mutation (8), vilken sedan kan nedärvas inom familjen. De vanligaste sjukdomsorsakande generna i Sverige är C9orf72HRE, SOD1 och FUS (7)(9)(10).
Andra etablerade riskfaktorer för ALS är hög ålder och manligt kön. Sannolika riskfaktorer inkluderar hyperlipidemi, rökning, måttlig till hög fysisk aktivitet, samt exponering för bly och pesticider (11)(12)(13).
Diagnoskriterier
Diagnosen ALS fastställs utifrån en kombination av kliniska fynd och noggrant uteslutande av relevanta differentialdiagnoser.
Gold Coast-kriterierna (14), som publicerades år 2020, underlättar att tidigt ställa diagnos och sätta in behandling. Kriterierna har hög sensitivitet, men det krävs erfarenhet för att använda dem korrekt och undvika feldiagnostisering.
För att ställa diagnos ALS enligt Gold Coast-kriterierna krävs:
- att patienten uppvisar progressiv muskelsvaghet samt tecken på både övre och nedre motorneuronsymtom i minst en kroppsregion, eller enbart nedre motorneuronsymtom i minst två kroppsregioner
- att differentialdiagnoser har uteslutits.
Observera att PLS inte omfattas av Gold Coast-kriterierna.
Sjukdomsförlopp
ALS kan debutera i mun och svalg, en arm eller ett ben, i bålen, i andningsmuskulaturen eller som kognitiv sjukdom (frontotemporal demens). Sjukdomen sprider sig successivt till fler muskler, vilket ofta leder till betydande fysisk funktionsnedsättning och psykiska påfrestningar för patient och närstående.
Exempel på typiska symtom som kan utvecklas när som helst under sjukdomsförloppet:
- bulbära symtom, till exempel talsvårigheter och sväljsvårigheter med hypersalivering
- muskelsvaghet i armar, ben, nacke eller bål
- andningspåverkan
- spasticitet och muskelkramper
- pseudobulbär affekt (affektlabilitet)
- ångest och depression
- kognitiva eller beteendemässiga funktionsnedsättningar.
Det finns ingen botande behandling mot ALS. Den genomsnittliga överlevnaden för en person med ALS är cirka tre år från symtomdebut, men det finns en mycket stor individuell variation. Personer med PSMA och PLS har långsammare sjukdomsprogress och har generellt en längre överlevnadstid.
Andningspåverkan
Andningspåverkan är vanligt vid ALS och orsakas i första hand av svaghet i andningsmuskulaturen. Det leder till underventilering med koldioxidretention och senare även nedsatt syremättnad, trots ofta friska lungor. Den progressiva andningssvikten med koldioxidretention, ibland i kombination med luftvägsinfektion, är den vanligaste dödsorsaken hos personer med ALS. Akut luftvägsobstruktion med mekanisk kvävning som direkt dödsorsak är mycket ovanlig (15).
Svag bukmuskulatur och nedsatt glottisfunktion minskar också hostkraften, vilket ökar risken för sekretstagnation och luftvägsinfektioner. Vid sväljsvårigheter ökar även risken för aspiration, vilket ytterligare kan bidra till luftvägsinfektioner och försämrat gasutbyte.
Fördjupad information om andning finns i Kliniskt kunskapsstöd ALS – handläggning vid andningspåverkan, 1177 för vårdpersonal.
Kognition och beteendeförändringar
Kognitiva och beteendemässiga funktionsnedsättningar kan observeras hos upp till 50 % av personer med ALS (16)(17). Dessa förändringar är i majoriteten av fallen lindriga och vanligen stabila över tid (18). Hos cirka 10 % av alla personer med ALS utvecklas däremot olika grader av frontotemporal demens. Dessa progredierar ofta över tid, vilket medför betydande funktionsnedsättning i vardagen (19).
Ingång och utgång
Ingång i vårdförloppet
Ingång i vårdförloppet sker vid misstanke om ALS hos en patient.
Misstanke om ALS väcks om progressiv muskelsvaghet noteras. Svagheten börjar ofta i en kroppsdel och sprider sig sedan med varierande hastighet. Beroende på vilken kroppsdel muskelsvagheten börjar benämns det olika:
- spinal start: debut i en arm, ett ben eller bål
- bulbär start: debut i mun och svalg.
Misstanke om ALS kan också väckas om det förekommer diffusa debutsymtom med exempelvis andningspåverkan, ofrivillig viktnedgång eller kognitiva och beteendemässiga funktionsnedsättningar. Vid frånvaro av muskelsvaghet eller andra typiska tecken på ALS, såsom fascikulationer, ligger fokus initialt på att utesluta differentialdiagnoser och ingången in i vårdförloppet fördröjs.
Utgång ur vårdförloppet
Utgång ur påbörjat vårdförlopp kan ske på något av följande sätt:
- misstanke om ALS avskrivs
- patienten avlider och efterlevandesamtal har erbjudits för att fånga upp närståendes behov av stöd.
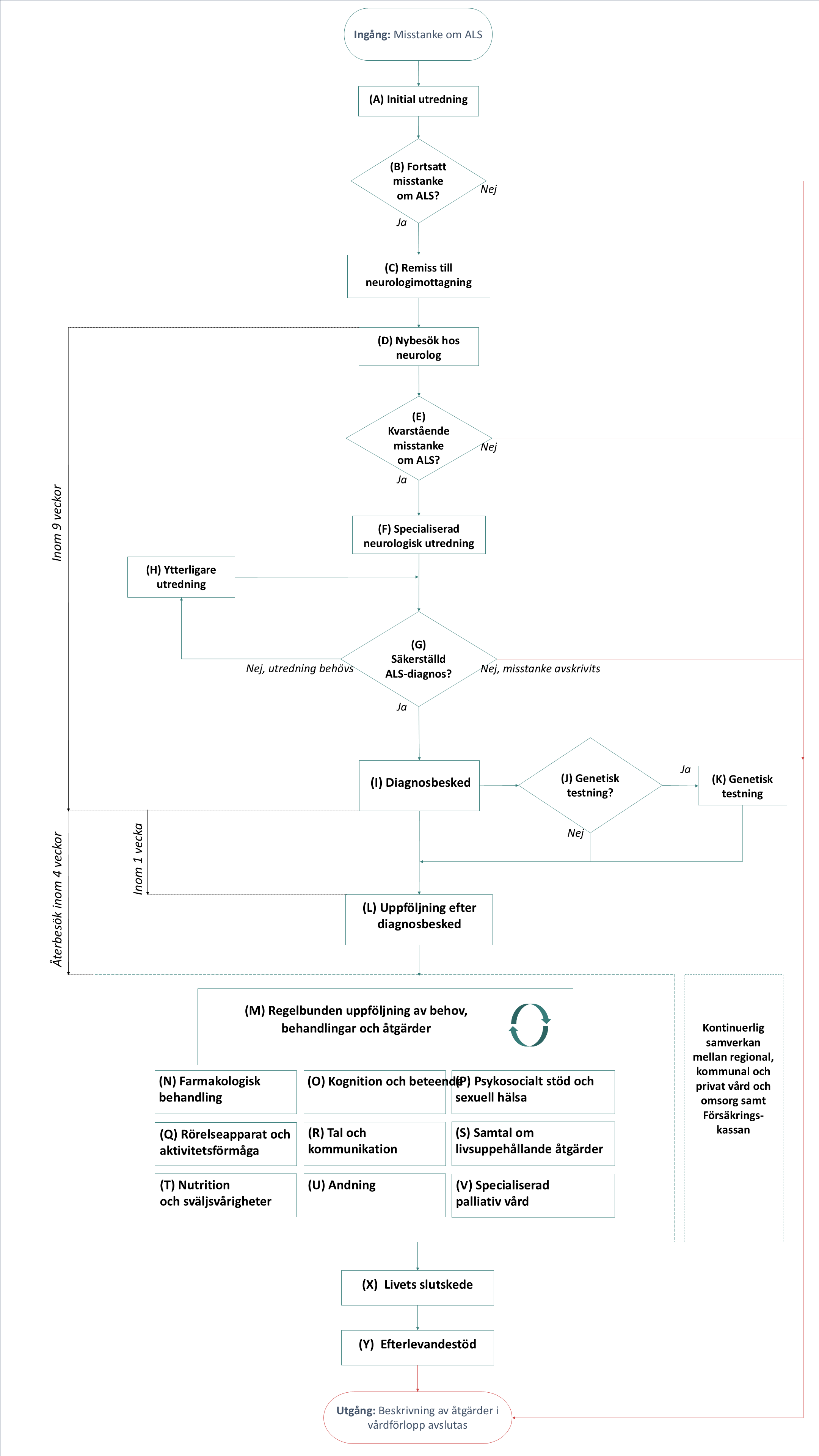
Flödesschema för vårdförloppet
Flödesschemat är en grafisk översikt av de åtgärder som ingår i vårdförloppet. Beskrivning i text finns i åtgärdsbeskrivningen.
* Utifrån symtom och behov bör relevanta vårdprofessioner kontaktas redan under åtgärderna [A-L].
Vårdförloppets åtgärder
Här beskrivs de åtgärder som ingår i vårdförloppet.
Patientmedverkan och kommunikation
Personcentrering och dokumenterad överenskommelse
Personcentrerad vård innebär att patientens och närståendes behov, erfarenheter och resurser är utgångspunkten för vården. Vården anpassas efter den enskilda patientens situation och värderingar, inom ramen för hälso- och sjukvårdens förutsättningar. En individuell prioritering av insatser är central för att skapa en god vårdallians med patienten och för att tidpunkten för olika samtal och åtgärder ska bli så ändamålsenlig som möjligt.
En personcentrerad vårdprocess bygger på en kontinuerlig dialog där vårdpersonalen uppmuntrar patienten att:
- reflektera över och uttrycka vad som är viktigt i livet och i vården
- kommunicera sin inställning till behandlingar och vårdbegränsningar
- vara delaktig i att identifiera lindrande strategier vid symtom.
För patienter med ALS, där sjukdomen successivt begränsar den fysiska och i vissa fall kognitiva förmågan, är en individuell vårdplan ett avgörande verktyg för att möjliggöra en personcentrerad, sammanhållen vård och skapa delaktighet i beslutsfattandet.
Eftersom ALS är en progressiv sjukdom behövs en individuell vårdplan. Vårdplanen uppdateras proaktivt i takt med förändrade behov, för att ge trygghet och förebygga akuta försämringar. Vårdplanen tydliggör vårdens innehåll, begränsningar och mål samt klargör ansvarsfördelning av planerade insatser.
Ytterligare aspekter vid upprättande av individuell vårdplan:
- Anpassa planen efter patientens livssituation, behov och önskemål.
- Inkludera närstående och annan vårdpersonal i planeringen.
- Beakta kulturella och existentiella värderingar om livets slut, med patientens samtycke.
- Säkerställ tillgång till planen för patient, närstående och vårdpersonal.
- Dokumentera i journalen:
- förebyggande insatser till exempel mot andnöd, smärta, infektioner
- åtgärder vid försämrade symtom
- beslut om vårdbegränsningar och symtomlindring
- önskemål om vårdplats och palliativ vård.
Utmaningar och mål
Patientens utmaningar
Utifrån enkäter och intervjuer med patienter och närstående för att fånga deras erfarenheter har följande övergripande utmaningar identifierats:
- Att patienter drar sig för att söka vård, då symtomen kan vara diffusa och det kan finnas en oro för att inte bli tagen på allvar.
- Att patienter kan känna oro och ångest när symtomen tilltar och förändras.
- Att patienter tycker det är jobbigt när utredningen drar ut på tiden samtidigt som oro för allvarlig diagnos finns.
- Att det kan vara svårt att ta till sig information och att det kan behövas psykosocialt och praktiskt stöd för patienten och närstående.
- Att det kan vara svårt att förstå roller och ansvarsfördelning mellan olika vårdaktörer och myndigheter samt att samordna de olika vårdaktörerna och myndigheterna som behöver involveras.
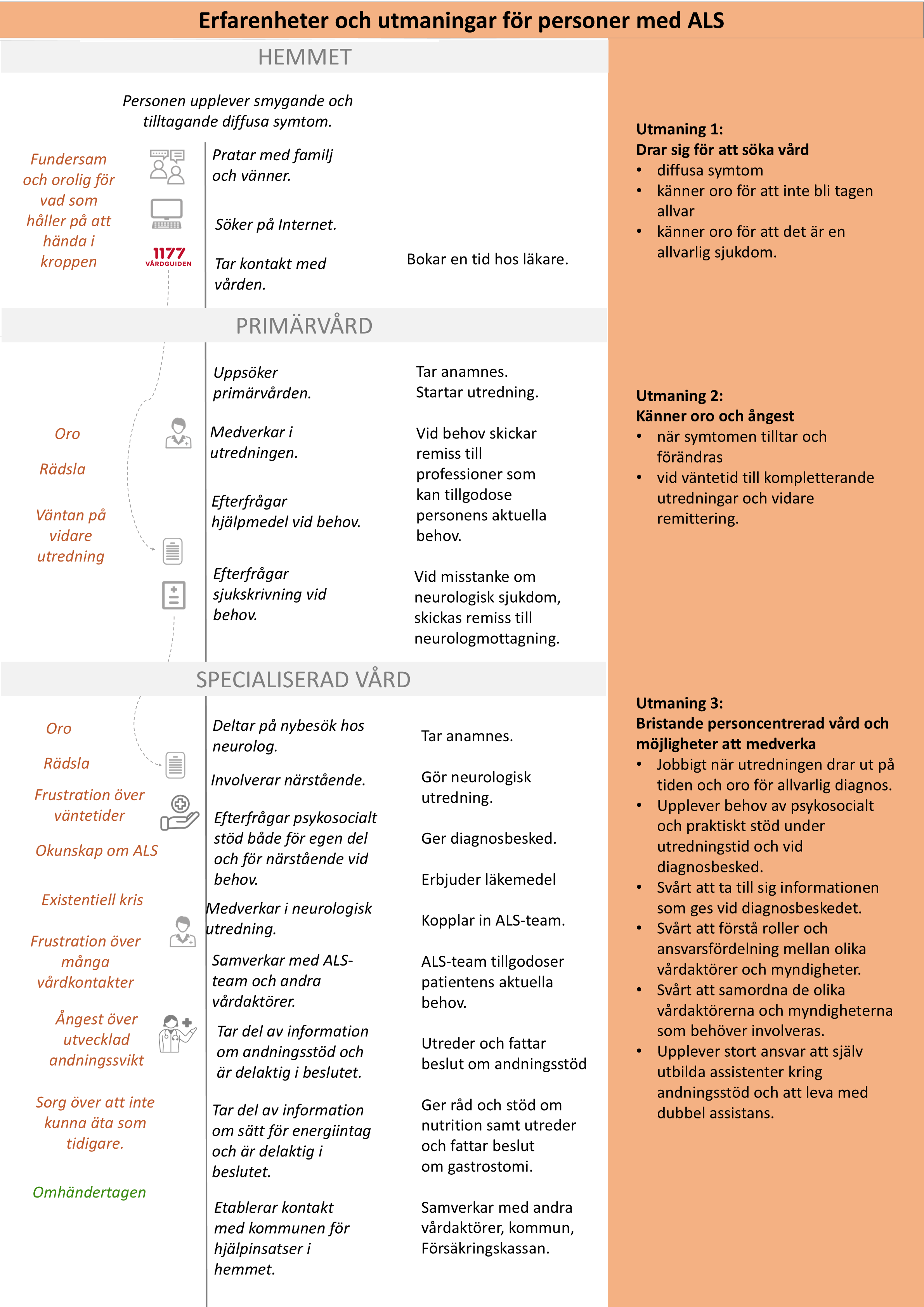
Nulägesbeskrivning av patienters erfarenheter
Nulägesbeskrivningen är en grafisk presentation av i nuläget vanligt förekommande erfarenheter av hälso- och sjukvården hos personer med ALS. Beskrivningen är framtagen utifrån intervjuer och enkätsvar från personer med ALS och deras närstående:
- Kolumn ett beskriver känslor och erfarenheter som patienterna berättar att de vanligtvis är med om.
- Kolumn två beskriver aktiviteter och åtgärder som patienten gör.
- Kolumn tre anger kort aktiviteter och åtgärder som hälso- och sjukvården gör.
- Kolumn fyra redovisar, utifrån patienternas perspektiv, de huvudsakliga utmaningar som patienterna berättar om. Vårdförloppet är utformat för att adressera dessa utmaningar, vilka även avspeglas i mål och indikatorer.
Vårdförloppets mål
Vårdförloppets mål är:
- Att alla patienter med ALS får diagnosbesked inom nio veckor från det att remissen skickades till neurolog.
- Förlängd överlevnadstid för patienter med ALS från diagnos.
- Att alla patienter med ALS känner sig delaktiga i beslut som rör deras sjukdom.
- Att alla patienter med ALS som befinner sig i livets slutskede erhåller adekvat symtomlindring.
- Ökad överlevnad och förbättrad hälsorelaterad livskvalitet för patienter med ALS genom att sätta in rätt resurser i rätt tid.
- Att beslut fattas om gastrostomisond, NIV, invasiv ventilation, och hjärt-lungräddning (HLR) för alla patienter med ALS inom tolv månader efter diagnos.
- Att alla patienter med ALS erbjuds regelbunden uppföljning av behov, behandling och åtgärder för att minska akuta insatser.
Kvalitetsuppföljning
Indikatorer för uppföljning
Indikatorerna i detta avsnitt visar vilka indikatorer som avses följas i vårdförloppet. I uppföljningsbilagan finns mer information om uppföljning av detta vårdförlopp och dess indikatorer.
Mer information om uppföljning av vårdförlopp finns under rubriken Generellt om personcentrerade och sammanhållna vårdförlopp.
Processmått
För att utvärdera vårdprocessen används följande mått:
- Andel patienter med ALS som får diagnosbesked inom nio veckor från det att remissen skickades till neurolog.
- Andel patienter med ALS som har tillgång till specialiserad palliativ vård i livets slutskede.
- Andel patienter med ALS som genomgår screening av andningsfunktionen inom två månader efter diagnos.
- Andel patienter med ALS med dokumenterad inställning till gastrostomisond, NIV, IV, och hjärt-lungräddning (HLR) inom tolv månader efter diagnos.
Resultatmått
För att utvärdera vårdens resultat används följande mått:
- Överlevnadstid (mediantid) för patienter med ALS från diagnos.
- Självskattad patientnöjdhet.
- Antal akuta inskrivningar vid sjukhus per patient och år.
I Bilaga 16 – Uppföljning av vårdförlopp ALS finns mer information om möjliga datakällor, uppföljning av vårdförloppet och dess indikatorer. Läs mer om uppföljning av vårdförlopp under rubriken Generellt om personcentrerade och sammanhållna vårdförlopp.
Kvalitetsregister
Rapportera gärna i följande kvalitetsregister (mer information finns även i åtgärdsbeskrivningen) oavsett om de används för uppföljning av vårdförloppets indikatorer eller inte:
- I ALS-registret, Svenska neuroregister rapporteras samtliga patienter och tänk på att:
- vid andningsscreening: ange datum för spirometri och resultatet
- vid andningsbehandling: registrera startdatum för NIV och IV
- övriga andningsvariabler kan, beroende på lokal rutin, i stället registreras i Swedevox.
- Swedevox är ett alternativt register för övriga andningsvariabler när lokal rutin så anger.
- I Svenska palliativregistret registreras samtliga dödsfall.
Sammanfattning av vårdförloppet
Vårdförloppet för amyotrofisk lateralskleros (ALS) beskriver en personcentrerad och sammanhållen vårdprocess från misstanke om ALS till dess att diagnosen avskrivs eller patienten avlider. Syftet är att bidra till jämlik vård, tydliga ansvar och strukturerade insatser genom hela sjukdomsförloppet. Vårdförloppet omfattar bland annat tidig diagnostik, multiprofessionellt omhändertagande och kontinuerlig uppföljning.
Vid misstanke om ALS remitteras patienten skyndsamt till neurolog för utredning. Målsättningen är att diagnosbesked ges och neuroprotektiv behandling startas inom nio veckor från att remiss skickats till neurolog. Diagnoskriterierna följer Gold Coast-kriterierna, och differentialdiagnoser utesluts systematiskt. Genetisk testning och vägledning erbjuds alla patienter.
Behandlingen syftar till symtomlindring och bibehållen livskvalitet. Farmakologiska och icke-farmakologiska insatser ges utifrån behov, exempelvis vid salivläckage, sekretstagnation och spasticitet. Nutrition och sväljförmåga följs strukturerat och gastrostomisond kan bli aktuell. Andningsfunktionen screenas och följs regelbundet och andningsstöd erbjuds vid behov.
ALS-vården kräver samverkan mellan en rad vårdaktörer. Involvera ett ALS-team tidigt som kan följa patienten kontinuerligt.
Palliativt förhållningssätt genomsyrar hela vården. Beslut om livsuppehållande åtgärder, som gastrostomisond, NIV och invasiv ventilation, tas i dialog med patienten och närstående. Specialiserad palliativ vård erbjuds vid komplexa vårdbehov och efterlevandestöd ges till närstående.
Vårdförloppet syftar till att minimera oönskad variation i vården och bygger på strukturerad kvalitetsuppföljning via kvalitetsregister. Personcentrering är en central del, där patientens behov och önskemål styr vårdplaneringen.
Ett kompletterande Kliniskt kunskapsstöd ALS – handläggning vid andningspåverkan har tagits fram och bör användas parallellt med detta vårdförlopp vid andningsrelaterade frågeställningar.
Generellt om personcentrerade och sammanhållna vårdförlopp
Om vårdförlopp
Personcentrerade och sammanhållna vårdförlopp syftar till att uppnå ökad jämlikhet, effektivitet och kvalitet i hälso- och sjukvården samt att skapa en mer välorganiserad och helhetsorienterad process för patienten.
Vårdförloppen utgår från tillförlitliga och aktuella kunskapsstöd och tas gemensamt fram av olika professioner inom regionernas nationella system för kunskapsstyrning.
I vårdförloppet beskrivs kortfattat vad som ska göras, i vilken ordning och när. Det beskriver en personcentrerad och sammanhållen vårdprocess som omfattar en hel eller en del av en vårdkedja. Åtgärderna kan individanpassas och inkluderar hur individens hälsa kan främjas.
Vårdförloppen avser minska oönskad variation och extra fokus riktas till det som inte fungerar i nuläget ur ett patientperspektiv. Det personcentrerade förhållnings- och arbetssättet konkretiseras genom patientkontrakt som tillämpas i vårdförloppen.
Den primära målgruppen för kunskapsstödet är hälso- och sjukvårdspersonal som ska få stöd i samband med vårdmötet. Avsnitten som rör utmaningar, mål och uppföljning av vårdförloppet är främst avsedda att användas tillsammans med beskrivningen av vårdförloppet vid införande, verksamhetsutveckling och uppföljning. De riktar sig därmed till en bredare målgrupp.
Om personcentrering
Ett personcentrerat förhållnings- och arbetssätt konkretiseras genom dokumenterad överenskommelse, som är en gemensam överenskommelse mellan vården och patienten om fortsatt vård och behandling.
Den dokumenterade överenskommelsen utgår från patientens och närståendes behov, resurser och erfarenheter av hälso- och sjukvård och innebär att en eller flera fasta vårdkontakter utses samt att det framgår vad vården tar ansvar för och vad patienten kan göra själv.
En dokumenterad överenskommelse kan göras vid flera tillfällen, relaterat till patientens hälsotillstånd.
Mer information finns på SKR:s webbsida om dokumenterad överenskommelse.
Om kvalitetsuppföljning
Vårdförloppen innehåller indikatorer för att följa upp i vilken grad patienten har fått vård enligt vårdförloppet. Befintliga källor används i den mån det går, men målsättningen är att strukturerad vårdinformation ska utgöra grunden för kvalitetsuppföljningen. Kvalitetsuppföljningen ska så stor utsträckning som möjligt baseras på information som är relevant för vården av patienten.
Vårdförloppets mål och åtgärder följs upp genom resultat- och processmått, vilket skapar förutsättningar för ett kontinuerligt förbättringsarbete.
För detaljerad information om hur indikatorerna beräknas, hänvisas till webbplatsen Kvalitetsindikatorkatalog där kompletta specifikationer publiceras i takt med att de är genomarbetade. Där beskrivs och motiveras också de valda indikatorerna.
Indikatorerna redovisas på Vården i siffror vartefter data finns tillgängligt. Data redovisas könsuppdelat och totalt, och för både region- och enhetsnivå när det är möjligt och relevant.
Relaterad information
Kunskapsstöd ALS, Socialstyrelsen
Palliativ vård, 1177 för vårdpersonal
Nationellt vårdprogram Palliativ vård, Palliativregistret
Webbutbildningen ALS - för personal inom kommunal vård och omsorg, Västra Götalandsregionen finns tillgänglig för alla i regionens digitala lärmiljö Lärportalen. Huvudsaklig målgrupp är vårdpersonal, personliga assistenter och hemtjänstpersonal inom kommunal och privat verksamhet, men kursen är fri att använda för alla. För att ta del av utbildningen, logga in som följande alternativ på webbplatsen Lärportalen VGR: Logga in på webbplatsen:
- Anställd inom VGR loggar in med VGR-id.
- Anställd hos en privat vårdgivare kopplad till VGR loggar in med annat konto och anger sitt VGR-id.
- Anställd inom kommun, region (som inte är Västra Götalandsregionen), universitet, övriga företag eller privatperson skapar ett konto för att logga in i Lärportalen.
Frågor om webbutbildningen? Kontakta uppdragsutbildning@vgregion.se.
Kompletterande underlag
Bilagor
Bilaga 1 – Initial utredning inför eventuell remiss till neurologimottagning
Bilaga 2 – Kliniska statusfynd vid besök hos neurolog
Bilaga 3 – Neurofysiologisk utredning
Bilaga 4 – Blod- och likvorprover
Bilaga 5 – Differentialdiagnoser
Bilaga 7 – Allmän palliativ vård
Bilaga 9 – Farmakologisk behandling
Bilaga 10 – Sexuell hälsa och reproduktion
Bilaga 12 – Rörelseapparaten och aktivitetsförmåga
(1) Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17085.
(2) Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942–55.
(3) Imrell S, Fang F, Ingre C, Sennfält S. Increased incidence of motor neuron disease in Sweden: a population-based study during 2002-2021. Journal of neurology. 2024;271(5):2730–5.
(4) Longinetti E, Regodon Wallin A, Samuelsson K, Press R, Zachau A, Ronnevi LO, et al. The Swedish motor neuron disease quality registry. Amyotrophic lateral sclerosis and frontotemporal degeneration. 2018;19(7-8):528–37.
(5) Svenska Neuroregister, ALS-registret [cited 2025- 08-09]. Available from: https://neuroreg.se/motorneuronsjukdom/om/.
(6) Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7(11):603–15.
(7) Andersen PM, Forsgren L, Binzer M, Nilsson P, Ala-Hurula V, Keränen ML, et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain. 1996;119 ( Pt 4):1153–72.
(8) Müller K, Oh KW, Nordin A, Panthi S, Kim SH, Nordin F, et al. De novo mutations in SOD1 are a cause of ALS. J Neurol Neurosurg Psychiatry. 2022;93(2):201–6.
(9) Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nature neuroscience. 2015;18(5):631–6.
(10) Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L, et al. The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur J Hum Genet. 2013;21(1):102–8.
(11) Goutman SA, Hardiman O, Al-Chalabi A, Chió A, Savelieff MG, Kiernan MC, et al. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022;21(5):465–79.
(12) Longinetti E, Fang F. Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Current opinion in neurology. 2019;32(5):771–6.
(13) Vasta R, Chia R, Traynor BJ, Chiò A. Unraveling the complex interplay between genes, environment, and climate in ALS. EBioMedicine. 2022;75:103795.
(14) Shefner JM, Al-Chalabi A, Baker MR, Cui LY, de Carvalho M, Eisen A, et al. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol. 2020;131(8):1975–8.
(15) Abbey E, Ali M, Cooper M, Taylor P, Mayland CR. Recognising dying in motor neurone disease: A scoping review. Palliat Med. 2024;38(9):923–34.
(16) Abrahams S. Neuropsychological impairment in amyotrophic lateral sclerosis-frontotemporal spectrum disorder. Nat Rev Neurol. 2023;19(11):655–67.
(17) Strong MJ, Abrahams S, Goldstein LH, Woolley S, McLaughlin P, Snowden J, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotrophic lateral sclerosis and frontotemporal degeneration. 2017;18(3-4):153–74.
(18) Finsel J, Uttner I, Vázquez Medrano CR, Ludolph AC, Lulé D. Cognition in the course of ALS-a meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. 2023;24(1-2):2–13.
(19) Antonioni A, Raho EM, Lopriore P, Pace AP, Latino RR, Assogna M, et al. Frontotemporal Dementia, Where Do We Stand? A Narrative Review. Int J Mol Sci. 2023;24(14).
(20) Socialstyrelsen. Socialstyrelsens termbank 2025 [cited 2025- 12-28]. Available from: https://termbank.socialstyrelsen.se/article.php?term=c2FtdGFsIHZpZCBhbGx2YXJsaWcgc2p1a2RvbQ==.
(21) Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, Van Damme P, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur J Neurol. 2012;19(3):360–75.
(22) NICE. Motor neurone disease: assessment and management 2016 [cited 2025- 04-22]. Available from: https://www.nice.org.uk/guidance/ng42.
(23) Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). Journal of the neurological sciences. 1999;169(1-2):13–21.
(24) Paynter C, Mathers S, Gregory H, Vogel AP, Cruice M. The impact of communication on healthcare involvement for people living with motor neurone disease and their carers: A longitudinal qualitative study. Int J Lang Commun Disord. 2022;57(6):1318–33.
(25) de Almeida FEO, do Carmo Santana AK, de Carvalho FO. Multidisciplinary care in Amyotrophic Lateral Sclerosis: a systematic review and meta-analysis. Neurol Sci. 2021;42(3):911–23.
(26) Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cognition and behaviour changes in ALS. Amyotrophic lateral sclerosis and frontotemporal degeneration. 2014;15(1-2):9–14.
(27) Vinceti G, Olney N, Mandelli ML, Spina S, Hubbard HI, Santos-Santos MA, et al. Primary progressive aphasia and the FTD-MND spectrum disorders: clinical, pathological, and neuroimaging correlates. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(3-4):146–58.
(28) Van Damme P, Al-Chalabi A, Andersen PM, Chiò A, Couratier P, De Carvalho M, et al. European Academy of Neurology (EAN) guideline on the management of amyotrophic lateral sclerosis in collaboration with European Reference Network for Neuromuscular Diseases (ERN EURO-NMD). Eur J Neurol. 2024;31(6):e16264.
(29) Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67(6):361–70.
(30) Carvalho TL, de Almeida LM, Lorega CM, Barata MF, Ferreira ML, de Brito-Marques PR, et al. Depression and anxiety in individuals with amyotrophic lateral sclerosis: a systematic review. Trends Psychiatry Psychother. 2016;38(1):1–5.
(31) Gould RL, McDermott CJ, Thompson BJ, Rawlinson CV, Bursnall M, Bradburn M, et al. Acceptance and Commitment Therapy plus usual care for improving quality of life in people with motor neuron disease (COMMEND): a multicentre, parallel, randomised controlled trial in the UK. Lancet. 2024;403(10442):2381–94.
(32) Heidari ME, Nadali J, Parouhan A, Azarafraz M, Tabatabai SM, Irvani SSN, et al. Prevalence of depression among amyotrophic lateral sclerosis (ALS) patients: A systematic review and meta-analysis. J Affect Disord. 2021;287:182–90.
(33) Oh J, An J, Park K, Park Y. Psychosocial interventions for people with amyotrophic lateral sclerosis and motor neuron disease and their caregivers: a scoping review. BMC Nurs. 2024;23(1):75.
(34) Hälso- och sjukvårdslag 2017:30 kapitel 5§7 (2017).
(35) Malmström N, Jakobsson Larsson B, Nilsson S, Öhlén J, Nygren I, Andersen PM, et al. Living with a parent with ALS - adolescents' need for professional support from the adolescents' and the parents' perspectives. Amyotroph Lateral Scler Frontotemporal Degener. 2023:1–9.
(36) Socialtjänstlagen (2001:453), (2001).
(37) Arvanitakis M, Gkolfakis P, Despott EJ, Ballarin A, Beyna T, Boeykens K, et al. Endoscopic management of enteral tubes in adult patients - Part 1: Definitions and indications. European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy. 2021;53(1):81–92.
(38) Teramoto S, Yoshida K, Hizawa N. Update on the pathogenesis and management of pneumonia in the elderly-roles of aspiration pneumonia. Respir Investig. 2015;53(5):178–84.
(39) Li Z, Kang H. Efficacy of non-pharmacological interventions for individuals with amyotrophic lateral sclerosis: systematic review and network meta-analysis of randomized control trials. Sci Rep. 2024;14(1):11365.
(40) Tomik B, Guiloff RJ. Dysarthria in amyotrophic lateral sclerosis: A review. Amyotroph Lateral Scler. 2010;11(1-2):4–15.
(41) Cave R, Bloch S. Voice banking for people living with motor neurone disease: Views and expectations. Int J Lang Commun Disord. 2021;56(1):116–29.
(42) Onesti E, Schettino I, Gori MC, Frasca V, Ceccanti M, Cambieri C, et al. Dysphagia in Amyotrophic Lateral Sclerosis: Impact on Patient Behavior, Diet Adaptation, and Riluzole Management. Front Neurol. 2017;8:94.
(43) Ortega O, Sakwinska O, Combremont S, Berger B, Sauser J, Parra C, et al. High prevalence of colonization of oral cavity by respiratory pathogens in frail older patients with oropharyngeal dysphagia. Neurogastroenterol Motil. 2015;27(12):1804–16.
(44) Gotesman RD, Lalonde E, McKim DA, Bourque PR, Warman-Chardon J, Zwicker J, et al. Laryngospasm in amyotrophic lateral sclerosis. Muscle Nerve. 2022;65(4):400–4.
(45) Janse van Mantgem MR, van Eijk RPA, van der Burgh HK, Tan HHG, Westeneng HJ, van Es MA, et al. Prognostic value of weight loss in patients with amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. 2020;91(8):867–75.
(46) Bjelica B, Bartels MB, Hesebeck-Brinckmann J, Petri S. Non-motor symptoms in patients with amyotrophic lateral sclerosis: current state and future directions. J Neurol. 2024;271(7):3953–77.
(47) Nationell Riktlinje om handläggning av perkutan endoskopisk gastrostomi (PEG) och andra perkutana nutritionssonder: Svensk gastroenterologisk förening; 2023 [cited 2025- 02-04]. Available from: https://svenskgastroenterologi.se/kunskap/handlaggning-av-perkutan-endoskopisk-gastrostomi-peg-och-andra-perkutana-nutritionssonder-nationell-riktlinje/.
(48) Palliativ vård - nationellt vårdprogram Kunskapsbanken; [cited 2024- 11-05]. Available from: https://kunskapsbanken.cancercentrum.se/globalassets/vara-uppdrag/rehabilitering-palliativ-vard/vardprogram/nationellt-vardprogram-palliativ-vard.pdf.
(49) Socialstyrelsen. Livsuppehållande behandling [cited 2024- 12-06]. Available from: https://www.socialstyrelsen.se/kunskapsstod-och-regler/regler-och-riktlinjer/foreskrifter-och-allmanna-rad/konsoliderade-foreskrifter/20117-om-livsuppehallande-behandling/.
(50) Socialstyrelsen. Nationella riktlinjer – Målnivåer. Palliativ vård i livets slutskede. Målnivåer för indikatorer 2017 [cited 2026- 02-10]. Available from: https://www.socialstyrelsen.se/contentassets/7b1bb76869534fdfb78ade500b1c8ed4/2017-10-22.pdf.
(51) Europe ECo. Guide to the quality and safety of organs for transplantation. 2022.