

Lungfibros - vuxna
Lungfibros - vuxna
Omfattning av kunskapsstödet
Vårdförloppet inleds vid symtom eller undersökningsfynd som kan vara förenliga med lungfibros hos en patient och pågår vanligen livet ut. En fullständig beskrivning av kriterier för omfattningen finns under rubriken Ingång och utgång.
Relaterade kunskapsstöd
Se Nationellt kliniskt kunskapsstöd (nationelltklinisktkunskapsstod.se) för följande relaterade kunskapsstöd:
- Personcentrerade och sammanhållna vårdförlopp för:
- kroniskt obstruktiv lungsjukdom
- obstruktiv sömnapné
- palliativ vård
- rehabilitering och försäkringsmedicinskt arbete
- reumatoid artrit
- hjärtsvikt.
- Kliniska kunskapsstöd för:
- sarkoidos
- andningsbesvär palliativ vård.
- Vårdriktlinje för:
-
- systemisk skleros.
Om hälsotillståndet
Definition
Lungfibros är ett samlingsbegrepp för tillstånd med ärromvandling av lungvävnad och ingår i gruppen interstitiell lungsjukdom (ILD) [1]. Vårdförlopp lungfibros inkluderar en heterogen grupp inom de interstitiella lungsjukdomarna, där det gemensamma är progredierande eller risk för progredierande fibrotisering av lungvävnad. Lungfibros är ett vardagligt och ospecifikt begrepp som används i kommunikation med allmänheten för att generellt beskriva fibrotiserande ILD.
Se bilaga kompletterande bakgrund om interstitiell lungsjukdom (ILD).
|
Förkortning/begrepp |
Beskrivning |
|
ILD |
Interstitiell lungsjukdom |
|
Lungfibros |
Beskriver ILD där delar av lungvävnaden omvandlats till ärrvävnad |
|
Fibros |
Onormal bindvävsbildning/ärrbildning, jämför lungfibros |
|
Reaktiv fibros |
Fibros av begränsad omfattning och resultat av känd extern påverkan |
|
Fibrotiserande lungsjukdom |
Fibros i lungvävnaden som riskerar att öka i omfattning |
|
IPF |
Idiopatisk lungfibros. Tillhör de fibrotiserande lungsjukdomarna |
|
UIP |
Radiologiskt mönster som ses vid IPF, och ibland vid annan fibrotiserande lungsjukdom |
|
PPF |
Progressiv lungfibros som uppfyller kriterier för försämringen. Tillhör de fibrotiserande lungsjukdomarna |
|
SSc |
Systemisk skleros |
|
RA |
Reumatoid artrit |
|
SLE |
Systemisk lupus erythematosus |
Interstitiell lungsjukdom (ILD)
Interstitiell lungsjukdom (interstitial lung disease, ILD) är ett begrepp som beskriver en heterogen grupp av lungsjukdomar [1]. ILD drabbar båda lungorna i varierande grad och omfattar ett stort antal tillstånd med ett brett spektrum av orsaker [2]. Vidare finns varierande kliniska och radiologiska manifestationer samt patologiska karakteristika, och därmed också varierande utfall [1, 3-6]. Eftersom de patologiska förändringar som drabbar patienten huvudsakligen förekommer i lungans bindväv (området mellan lungcellerna, även kallat interstitium), kallas denna grupp av sjukdomar för interstitiella lungsjukdomar (ILD) [1].
ILD där det förekommer ärromvandling av lungparenkymet är fibrotisk och benämns i allmänhet ofta ”lungfibros” [2]. ILD som är icke-fibrotisk [7] kan läka ut, ibland av sig självt men ibland med hjälp av (immunmodulerande) läkemedelsbehandling, men kan även i vissa fall övergå i fibrotiserande lungsjukdom. Lungfibros utvecklas inte hos alla patienter med ILD. Det finns i dag inte några metoder för att avgöra om en patient kommer att utveckla lungfibros, men vissa tillstånd innebär större risk (se riskfaktorer) [1, 3-6].
Se bilaga kompletterande bakgrund om interstitiell lungsjukdom (ILD).
Lungfibros
Begreppet lungfibros beskriver tillståndet där delar av lungvävnaden omvandlats till ärrvävnad [1, 2, 7]. Det kan ske vid flera sjukdomar och tillstånd som ryms under det övergripande begreppet interstitiell lungsjukdom (ILD) [1]. Fibrotisering är en skada som inte är reversibel. Vid lungfibros som orsakats av specifik exponering, exempelvis asbestos, kan den avstanna efter att orsaken avlägsnats. Fibrotisering som inte avstannar benämns progressiv och innebär att en allt större del av patientens lungvävnad successivt fibrotiseras [1, 2, 7].
Termen "fibros" betyder onormal bindvävsbildning/ärrbildning. Det är en strikt beskrivande term som inte innehåller uppgifter om bakomliggande orsak. När den onormala bindvävsbildningen/ärrbildningen sker i lungvävnaden används ofta termen ”lungfibros”. På samma sätt som termen "fibros" är den enbart beskrivande och innehåller inga uppgifter om sin bakomliggande orsak [1].
Lungfibros kan ha många orsaker och kan delas in i två huvudkategorier [1-3, 7]:
- Reaktiv fibros: Fibros som är av begränsad omfattning och resultat av känd extern påverkan. Ett exempel på detta är fibros som kan komma efter strålbehandling eller efter svåra lung- och lungsäcksinfektioner.
- Fibrotiserande lungsjukdom:Fibros i lungvävnaden som riskerar att progrediera (öka i omfattning). Det finns ett antal olika former med varierande prognos och förlopp, den mest välkända är idiopatisk lungfibros (idiopathic pulmonary fibrosis, IPF).
Termen "lungfibros" har sedan flera år använts som ett övergripande begrepp för såväl reaktiva förändringar som för fibrotiserande lungsjukdomar. De senaste decennierna har de fibrotiserande lungsjukdomarna kommit i fokus med ökad kunskap om behandling och prognos. Det har uppstått ett behov att differentiera mellan dessa två kategorier av fibros, dels för att bespara patienterna onödig oro, dels för att kunna fokusera på den kategori patienter som har större behov av omhändertagande och behandling [2, 7].
Idiopatisk lungfibros (IPF)
De fibrotiserande lungsjukdomarna kan delas in i specifika medicinska diagnoser. IPF är den fibrotiserande lungsjukdom som är mest beskriven och karakteriserad [3, 8-10]. Utmärkande för IPF är att diagnosen ställs genom usual interstitial pneumonia (UIP)-mönster tillsammans med klinisk bild. UIP-mönster kan förekomma på [8]:
- högupplöst datortomografi (high resolution computed tomography, HRCT) (se diagnoskriterier)
- datortomografi (DT) vid utbredd fibrotisering
- vävnadsprov (biopsi) från lungan.
IPF är bland de ILD-sjukdomar med sämst prognos. Utan sjukdomsspecifika läkemedel beräknas, i olika studier, överlevnaden till mellan tre och fem år från diagnos, variation förekommer [3, 4, 8, 11]. Detta vårdförlopp refererar därför ibland till erfarenheter från specifikt IPF men konsensus är att det är applicerbart även på övrig progressiv fibrotiserande lungsjukdom [1].
Progressiv lungfibros (PPF)
Progressiv lungfibros (progressive pulmonary fibrosis, PPF) är en allmän definition för patienter med progressiv fibrotiserande lungsjukdom, annan än IPF, av känd eller okänd bakomliggande orsak [3].
ILD vid autoimmun sjukdom
ILD med risk för, eller utvecklad, fibrotiserande lungsjukdom förekommer i hög men varierande frekvens vid flera autoimmuna sjukdomar [2, 5, 12], framför allt hos patienter med:
- systemisk skleros (SSc)
- inflammatorisk idiopatisk myopati (myosit)
- reumatoid artrit (RA).
Det förekommer även vid följande tillstånd:
- Sjögrens syndrom
- systemisk lupus erythematosus (SLE)
- andra inflammatoriska systemsjukdomar.
Förekomst
Interstitiell lungsjukdom (ILD) - Prevalensen för ILD är 6—76/100 000 [1, 2, 8, 11, 13, 14].
Idiopatisk lungfibros (IPF) - IPF är den variant av fibrotiserande lungsjukdom som är bäst beskriven vad gäller prevalens [8, 15]. I Finland beräknades den till 1–28/100 000 med en incidens på 1–9/100 000 per år [8]. En översikt från Tyskland visar en punktprevalens på strax ovan 20/100 000 och en incidens på strax ovan 10/100 000, per år [9]. Svenska siffror är osäkra men tyder på liknande resultat. IPF är associerat till manligt kön och rökning [8, 11, 15].
ILD vid autoimmun sjukdom - ILD kan detekteras hos 30-70 % av patienter med systemisk skleros, 30—60 % av patienter med RA och upp till 80 % av dem med myosit [12-14, 16].
Orsaker
Lungfibros uppstår efter mikroskopisk skada i alveolärepitelet som inte läkt korrekt utan orsakat ärrbildning och därigenom sämre förmåga för syrgasutbyte [1]. Kända orsaker kan vara [1, 6, 17]:
- exponering för till exempel stendamm, asbest, strålbehandling
- autoimmun sjukdom
- läkemedel.
Lungfibros av okänd orsak benämns idiopatisk, exempelvis IPF (idiopatisk lungfibros).
Riskfaktorer
Ökad risk för lungfibros finns hos personer med [7, 10, 18]:
- pågående eller tidigare rökning
- exponering för partiklar
- interstitiella lungabnormaliteter (ILA).
Diagnoskriterier
Diagnosticering av lungfibros baseras i första hand på radiologiskt mönster från HRCT tillsammans med den kliniska bilden (anamnes, status samt förlopp) [2-4, 7, 8, 10, 19 ].
- Se åtgärd (A) samlad bedömning av anamnes, status och utredningsfynd.
- Se bilagor anamnes samt klinisk undersökning och utredning.
Bronkoskopi med bronkoalveolärt lavage (BAL) och/eller lungbiopsi kan bli aktuell om diagnos inte kan ställas med ovanstående underlag [3, 7, 8, 10].
Radiologi
Den radiologiska bilden i form av HRCT är en hörnsten i diagnosticering av ILD och särskilt fibrotiserande lungsjukdom [2-4, 7, 8, 10, 19]. Se även åtgärd (C).
Vid IPF kallas det radiologiska mönstret usual interstitial pneumonia (UIP) [7, 8, 19]. Mönstret kan även förekomma vid andra fibrotiserande lungsjukdomar, exempelvis vid fibrotiserande lungsjukdom associerad till RA [2].
Non-specific interstitial pneumonia (NSIP) är det mönster som ses vid sjukdomen idiopatisk NSIP (iNSIP) men även vid lungfibros associerad till autoimmun sjukdom. Organiserande pneumoni (OP) kan vara idiopatisk eller associerad till bland annat autoimmuna sjukdomar, infektioner och läkemedel [2, 4-6, 16].
Lungfunktionsmätning
Dynamisk spirometri kan väcka misstanke om restriktivitet men det är med statisk spirometri som sänkta lungvolymer säkerställs. Vid lungfibros finns ofta en restriktiv bild mätt med dynamisk och statisk spirometri samt nedsatt diffusionskapacitet för kolmonoxid (DLCO). Vid en blandbild av emfysem och fibros kan den dynamiska och statiska spirometrin vara helt normal men då är diffusionskapaciteten påverkad. [1-3, 7, 8, 10]
Se även artikel i European respiratory journal från 2022, ERS/ATS technical standard on interpretive strategies for routine lung function tests.
Multidisciplinär konferens (MDK)
Multidisciplinär konferens (MDK) bör skiljas från en vanlig röntgenrond. I MDK deltar [2-4, 8]:
- lungspecialister
- thoraxradiolog alternativt ILD-erfaren radiolog.
Samt utifrån behov:
- reumatolog
- klinisk fysiolog
- patolog
- thoraxkirurg
- kardiolog.
Dessa analyserar i samverkan olika aspekter av patientens besvär och undersökningsresultat, och kan därigenom diagnosticera ILD-sjukdomen samt ge förslag på fortsatt omhändertagande och behandling [2-4, 8].
Diagnostisering av progressiv lungfibros (PPF)
Radiologisk bild av fibrotiserande lungsjukdom och minst två av följande tre kriterier som inträffat under det senaste året utan annan alternativ förklaring till försämring [3]:
- förvärrade lungsymtom (hosta, andfåddhet)
- försämring av lungfunktion (mätt med FVC och DLCOc)
- progress av radiologiska förändringar.
Se åtgärd (K).
Fibrotiserande lungsjukdom vid autoimmun sjukdom
Vid progressiv fibrotiserande lungsjukdom associerad till autoimmun sjukdom, trots antireumatisk behandling, föreslås MDK där reumatolog, lungmedicinspecialist, thorax- /ILD-erfaren radiolog och vid behov klinisk fysiolog deltar [2-7, 16].
Vid både systemisk skleros och myosit ingår radiologisk utredning med HRCT och lungfunktionsundersökning inklusive mätning av DLCOc i den basala utredningen. Lungröntgen ingår i den initiala handläggningen vid RA, med kompletterande HRCT vid avvikande fynd.
Samsjuklighet
Tillstånd som kan förekomma samtidigt som ILD med eller utan lungfibros är reflux, astma, kroniskt obstruktiv lungsjukdom (KOL), hjärtsvikt, ångest samt post-infektiöst tillstånd [8, 17].
Tillstånd med ökad förekomst hos patienter med IPF jämfört med övrig population [8]:
- gastroesofageal reflux (GERD)
- obstruktiv sömnapné
- pulmonell hypertension
- kranskärlssjukdom
- kroniskt obstruktiv lungsjukdom (KOL)
- ångest/depression
- diabetes
- hjärtsvikt
- lungcancer
- lungemboli.
Sjukdomsförlopp
Dyspné, torrhosta och fatigue (trötthet) är initiala symtom, men är ospecifika för ILD inklusive lungfibros. Det är därför viktigt att utredning av sådana symtom inte avstannar utan att orsak identifieras och eventuella differentialdiagnoser utesluts [2-4, 8, 10, 18].
Från de första symtomen på ILD kan sjukdomen utvecklas på olika sätt beroende på om fibrotisering av lungvävnaden uppstår eller ej. Eftersom fibrotiserande lungsjukdom orsakar permanent lungfunktionsnedsättning innebär det därför generellt fler och mer uttalade svårigheter för patienten [1-5, 7, 8, 10, 12, 13, 17, 18]. Nedsatt ork och ökad dyspné begränsar aktiviteter i dagliga livet, därför är det av stor vikt att patienten får kunskap om och förstår hur hen kan hantera sjukdomens symptom i vardagen och på så vis möjliggöra att fortsätta utföra de aktiviteter som upplevs som meningsfulla [20, 21].
Progressiv fibrotiserande lungsjukdom och IPF orsakar en kontinuerlig förlust av lungfunktion med påföljande besvär hos patienterna så som [3, 8, 21, 22]:
- tilltagande dyspné
- torrhosta
- fatigue
- undernäring
- nedstämdhet
- begränsad möjlighet till fysisk aktivitet och vardagsaktiviteter
- sänkt hälsorelaterad livskvalitet
- minskad förväntad livslängd.
Insatser utifrån interprofessionell samverkan är av stor vikt [2, 8, 23]. Tillsammans med patienten består teamet av:
- sjuksköterska
- läkare
- fysioterapeut
- arbetsterapeut
- dietist
- kurator.
Tidiga rehabiliteringsinsatser kan förbättra fysisk kapacitet, öka patientens livskvalitet, minska dyspné och öka förmåga till fysisk aktivitet och träning, samt arbetsförmåga. Kartläggning av patientens dagliga liv och energibesparande strategier kan öka delaktighet i personens dagliga aktiviteter, samt öka kunskap att hantera symptom som nedsatt ork och dyspné [2, 20, 22, 24-26]. Lindrande vård inklusive syrgasbehandling är aktuellt att adressera tidigt i förloppet. Syrgasbehandling kan ges vid aktivitet och/eller vila, se åtgärd (M) [23, 27].
Se åtgärd (H) rehabilitering och hälsofrämjande åtgärder, samt generisk modell för rehabilitering (pdf).
Lungfunktion
Sjukdomsprogress vid lungfibros kan monitoreras med olika tester [3]. Testerna görs vanligtvis vid diagnostillfället, var tredje till sjätte månad under första året, och upprepas var sjätte till tolfte månad eller tätare vid behov [3, 8]. Följande tester utförs för uppföljning och bedömning av sjukdomsprogression:
- forcerad vitalkapacitet (FVC) som bestäms vid dynamisk spirometri
- diffusionskapacitet för kolmonoxid korrigerat för aktuellt Hb-värde (DLCOc).
Absolut minskning av FVC ≥ 5 % och/eller DLCOc ≥ 10 % under ett år anses vara tecken på sjukdomsprogress [3].
Funktionell fysisk förmåga
Påverkan från fibrotiserande lungsjukdom på fysisk förmåga kan monitoreras med olika tester [8, 20, 26, 28]. Sex minuters gångtest (6MGT) görs vanligtvis vid diagnostillfället och upprepas var tredje till sjätte månad samt vid behov. Det genomförs med bedömning av desaturation samt skattning av dyspné och bentrötthet. Tillförlitlig metod för att mäta saturation (pulsoximeter och/eller prob) används utifrån patientens förutsättningar. Patientens förväntade förvärde i procent kan räknas ut för att bedöma den fysiska kapaciteten [29].
Testet 1-minute sit-to-stand (1STS) kan också användas för att bedöma den funktionella muskelstyrkan i nedre extremiteten [26, 28]. Det genomförs med bedömning av desaturation samt skattning av dyspné och bentrötthet. Liksom vid 6MGT går det att räkna ut procent av förväntat värde [30]. Det är viktigt att individuellt anpassa testen efter patientens behov och att välja det som patienten kan utföra.
För yngre patienter kan en ergospirometri vara ett bra alternativ av test av fysisk kapacitet för att identifiera lättare funktionsnedsättning i ett tidigt skede.
Exacerbationer
Akut exacerbation (AE) vid fibrotiserande lungsjukdom definieras som en försämring som börjat under de senaste 30 dagarna utan annan förklaring. Det är viktigt att utesluta andra tillstånd som förklaring till försämringen (exempelvis akut försämring av hjärtsvikt, infektioner och lungemboli) och behandla dessa [2, 3, 8, 27].
Hälsorelaterad livskvalitet (HRQL)
HRQL, mätt med sjukdomsspecifika frågeformulär, tenderar att vara sänkt hos personer med fibrotiserande lungsjukdom [2, 8, 21]. Bidragande orsaker är begränsningar i det sociala livet och begränsningar av fysisk aktivitet inklusive nedsatt arbetsförmåga. God förmåga till egenvård, kunskap om sjukdomen och behandlingar, aktivt deltagande i sin vård samt stöd från närstående är faktorer som kan bidra till stabil eller ökad HRQL.
I Sverige mäts HRQL främst med Kings korta frågeformulär för lungfibros (K-BILD) [31], se lungfibrosregistret (slmf.se).
Medelvärde vid diagnostillfället bland patienter med IPF, mätt med K-BILD, är 54 poäng, vilket är en måttligt sänkt HRQL.
Ingång och utgång
Ingång i vårdförloppet
Ingång i vårdförloppet sker:
- när en person uppvisar symtom (vanligen ansträngningsutlöst dyspné eller torrhosta) och kliniska fynd (krepitationer och/eller desaturation vid ansträngning) som kan vara förenliga med ILD med eller utan fibros, och där infektion (pågående eller postinfektiöst tillstånd), hjärtsjukdom (framför allt hjärtsvikt), astma, kroniskt obstruktiv lungsjukdom (KOL) och lungcancer, inte är primär misstanke.
- när en patient med autoimmun sjukdom uppvisar symtom eller undersökningsfynd som kan bero på ILD
- när ILD som upptäcks som bifynd vid radiologisk undersökning.
Ingång kan ske oavsett vårdnivå.
Utgång ur vårdförloppet
Utgång ur vårdförloppet sker:
- om utredning visar att lungfibros inte föreligger
- vid lungtransplantation.
Flödesschema för vårdförloppet
Flödesschemat i Figur 1 beskriver de åtgärder som ingår i vårdförloppet. Beskrivning i text finns i åtgärdstabellen.
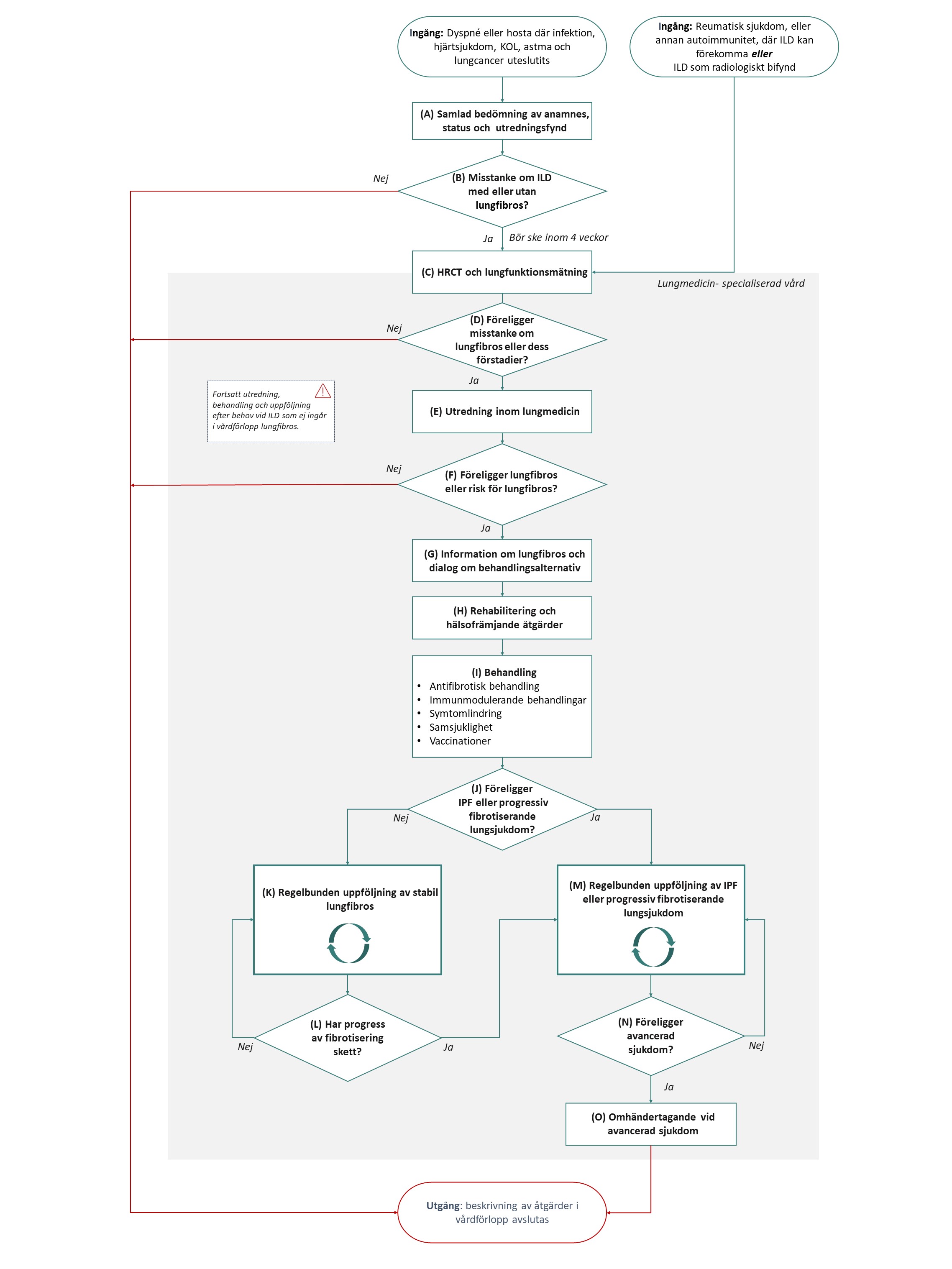
Vårdförloppets åtgärder
Här beskrivs de åtgärder som ingår i vårdförloppet.
Patientmedverkan och kommunikation
Personcentrering och dokumenterad överenskommelse
Personcentrering är beaktad i ovan beskrivna åtgärder genom att det är skrivet ur den drabbade patientens synvinkel och i varje del med den drabbade personens berättelse som grund. Utöver detta är nedanstående viktigt att lyfta fram.
Dokumenterad överenskommelse/patientkontrakt utformas gemensamt av patienten och dess fasta vårdkontakt och är en överenskommelse om planerad uppföljning, behandling samt kontaktuppgifter till vården. Det framgår också vem som är patientens fasta vårdkontakt. Där framgår när patienten kan förvänta sig nästa kontakt (utifrån personens behov) samt vid vilka symtom patienten ska höra av sig till vården och vart patienten kan vända sig. Det ska vara tydligt vilka åtgärder som vården ansvarar för och vad patienten själv kan göra. Det framgår även vilket stöd patienten kan få i form av interprofessionell samverkan samt vilka möjligheter till behandling som finns och vilka effekter, önskade och oönskade, dessa kan innebära. Patientkontraktet följs upp enligt åtgärder i detta vårdförlopp.
Utmaningar och mål
Patientens utmaningar
Utifrån patienters och närståendes erfarenheter har följande övergripande utmaningar identifierats:
- lång tid till påbörjad utredning av lungfibros
- avsaknad av personcentrerad vård och begriplig information under utredning och diagnostisering
- ojämlik vård
- lång tid till diagnosticering och behandling
- sänkt livskvalitet.
Nulägesbeskrivning av patienters erfarenheter
Figur 2 nedan är en grafisk presentation av i nuläget vanligt förekommande erfarenheter av hälso- och sjukvården hos vuxna med lungfibros.
- I kolumn 1 beskrivs identifierade positiva och negativa patientupplevelser.
- I kolumn 2 anges för patienten vanligt förkommande aktiviteter och åtgärder.
- I kolumn 3 beskrivs vårdens vanligt förekommande aktiviteter och åtgärder.
- I kolumn 4 beskrivs huvudsakliga utmaningar som patienten möter. Vårdförloppet är utformat för att adressera dessa utmaningar som även avspeglas i vårdförloppets mål och indikatorer.

Vårdförloppets mål
Vårdförloppets mål är att:
- Patienten genomgår en aktiv och skyndsam utredning tills orsaken till patientens initiala symtom har fastställts.
- Patientfallet tas upp på Multidisciplinär konferens (MDK):
- när läkare inom lungmedicinsk specialiserad vård, med stöd av thoraxradiologs utlåtande, inte kan fastställa diagnos
- om osäkerhet kring behandling föreligger
- för att säkerställa att en tydlig planering för vidare utredning och åtgärder finns, särskilt i de fall när diagnosticering är komplicerad.
- Patienten har tillgång till kontaktperson, kontinuerlig information, stöd och utbildning utifrån utredning, diagnos, behandling, rehabilitering i interprofessionell samverkan, hälsofrämjande åtgärder samt uppföljning och palliativ vård när behov uppstår.
Kvalitetsuppföljning
Vårdförloppets indikatorer presenteras här och i bilagan Uppföljning av vårdförlopp lungfibros finns mer information om uppföljning av vårdförloppet och dess indikatorer.
Läs mer om uppföljning av vårdförlopp under rubriken Generellt om personcentrerade och sammanhållna vårdförlopp.
Indikatorer för uppföljning
Resultatmått
- Andel patienter med IPF eller PPF med stabil eller ökad HRQL mätt med Kings korta frågeformulär för lungfibros (K-BILD) under 6 månader (andel utan minskning av totalpoäng ≥5)
- Andel patienter med IPF eller PPF med bibehållen eller ökad (≥30 meter) gångsträcka vid 6 minuters gångtest under 6 månader.
Processmått
- Genomsnittligt antal dagar från senaste utförda HRCT till diagnos för patienter med IPF eller PPF
- Genomsnittligt antal dagar till försämrad (minskad totalpoäng ≥5) HRQL, mätt med K-BILD beräknat från datum för diagnos för patienter med IPF eller PPF
- Genomsnittligt antal dagar till försämrad gångsträcka (minskad ≥30 meter) vid 6 minuters gångtest beräknat från datum för diagnos för patienter med IPF eller PPF
- Andel patienter som vid vårdkontakt får diagnosen IPF eller PPF för första gången och samtidigt får undersökningarna, 6 minuters gångtest, lungfunktion och sjukdomsspecifik HRQL
- Andel patienter med diagnosen IPF eller PPF som fått patientutbildning inom 12 månader från diagnos
- Andel patienter med diagnosen IPF eller PPF som fått en vårdplan inom 3 månader från diagnos
Bilaga Lungfibros vardforlopp.pdf
Kvalitetsregister
Följande kvalitetsregister kan vara relevanta att registrera i under någon del av vårdförloppet (se även åtgärdsbeskrivningen) oavsett om de används för uppföljning av vårdförloppets indikatorer eller inte.
Lungfibrosregistret samlar sedan 2014 information om personer med IPF i hela Sverige. Täckningsgrad är cirka 65 %. Resultatmått för gångsträcka (6MGT), lungfunktion samt HRQL (mätt med K-BILD) är exempel på mått som kan fås från registret. Det finns möjlighet att utöka registret till att inkludera alla personer med lungfibros.
Sammanfattning av vårdförloppet
Lungfibros är ett samlingsbegrepp för tillstånd med ärromvandling av lungvävnad och ingår bland de interstitiella lungsjukdomarna (interstitial lung diseases, ILDs). Diagnosticering av lungfibros baseras i första hand på radiologiskt mönster från högupplöst datortomografi (high resolution computed tomography, HRCT) tillsammans med den kliniska bilden (anamnes, status samt förlopp). ILD, och särskilt lungfibros, är ovanliga tillstånd, inte fler än ungefär 70 av 100 000 personer drabbas. Tillstånd som kan likna ILD-sjukdom med eller utan lungfibros är reflux, astma, kronisk obstruktiv lungsjukdom (KOL), hjärtsvikt, ångest samt post-infektiöst tillstånd.
Lungfibros i form av progressiv fibrotiserande lungsjukdom orsakar en kontinuerlig förlust av lungfunktion med påföljande besvär så som tilltagande dyspné, torrhosta, fatigue, undernäring, nedstämdhet, begränsad möjlighet till aktivitet, sänkt hälsorelaterad livskvalitet och minskad förväntad livslängd.
Vårdförloppet omfattar vuxna personer med lungfibros och inleds när en person uppvisar ansträngningsutlöst dyspné eller torrhosta samt krepitationer eller desaturation vid ansträngning och där infektion, hjärtsjukdom, astma, KOL och lungcancer uteslutits. Vårdförloppet kan även inledas genom att en patient med autoimmun sjukdom uppvisar symtom eller undersökningsfynd som kan bero på ILD, eller när ILD upptäcks som bifynd vid radiologisk undersökning. Åtgärderna avslutas om misstanke om lungfibros avskrivs eller om personen genomgår lungtransplantation.
Målet med vårdförloppet är att personer med lungfibros får möjlighet till utredning, behandling och omhändertagande utan onödiga dröjsmål. Målen är även att de får tillgång till kontaktperson, kontinuerlig information, stöd, utbildning, behandling, rehabilitering i interprofessionell samverkan, samt uppföljning och palliativ vård när behov uppstår. Uppföljning av vårdförloppet sker genom resultat- och processmått.
Vårdförloppet beskriver de åtgärder som utförs från den initiala utredningen, via djupare utredning efter stärkt misstanke om lungfibros, till diagnosticering, behandling och omhändertagande. Även skillnad i åtgärder vid stabil respektive progressiv sjukdom beskrivs.
Generellt om personcentrerade och sammanhållna vårdförlopp
Om vårdförlopp
Personcentrerade och sammanhållna vårdförlopp syftar till att uppnå ökad jämlikhet, effektivitet och kvalitet i hälso- och sjukvården samt att skapa en mer välorganiserad och helhetsorienterad process för patienten.
Vårdförloppen utgår från tillförlitliga och aktuella kunskapsstöd och tas gemensamt fram av olika professioner inom regionernas nationella system för kunskapsstyrning.
I vårdförloppet beskrivs kortfattat vad som ska göras, i vilken ordning och när. Det beskriver en personcentrerad och sammanhållen vårdprocess som omfattar en hel eller en del av en vårdkedja. Åtgärderna kan individanpassas och inkluderar hur individens hälsa kan främjas.
Vårdförloppen avser minska oönskad variation och extra fokus riktas till det som inte fungerar i nuläget ur ett patientperspektiv. Det personcentrerade förhållnings- och arbetssättet konkretiseras genom patientkontrakt som tillämpas i vårdförloppen.
Den primära målgruppen för kunskapsstödet är hälso- och sjukvårdspersonal som ska få stöd i samband med vårdmötet. Avsnitten som rör utmaningar, mål och uppföljning av vårdförloppet är främst avsedda att användas tillsammans med beskrivningen av vårdförloppet vid införande, verksamhetsutveckling och uppföljning. De riktar sig därmed till en bredare målgrupp.
Om personcentrering
Ett personcentrerat förhållnings- och arbetssätt konkretiseras genom dokumenterad överenskommelse, som är en gemensam överenskommelse mellan vården och patienten om fortsatt vård och behandling.
Den dokumenterade överenskommelsen utgår från patientens och närståendes behov, resurser och erfarenheter av hälso- och sjukvård och innebär att en eller flera fasta vårdkontakter utses samt att det framgår vad vården tar ansvar för och vad patienten kan göra själv.
En dokumenterad överenskommelse kan göras vid flera tillfällen, relaterat till patientens hälsotillstånd.
Mer information finns på SKR:s webbsida om dokumenterad överenskommelse.
Om kvalitetsuppföljning
Vårdförloppen innehåller indikatorer för att följa upp i vilken grad patienten har fått vård enligt vårdförloppet. Befintliga källor används i den mån det går, men målsättningen är att strukturerad vårdinformation ska utgöra grunden för kvalitetsuppföljningen. Kvalitetsuppföljningen ska så stor utsträckning som möjligt baseras på information som är relevant för vården av patienten.
Vårdförloppets mål och åtgärder följs upp genom resultat- och processmått, vilket skapar förutsättningar för ett kontinuerligt förbättringsarbete.
För detaljerad information om hur indikatorerna beräknas, hänvisas till webbplatsen Kvalitetsindikatorkatalog där kompletta specifikationer publiceras i takt med att de är genomarbetade. Där beskrivs och motiveras också de valda indikatorerna.
Indikatorerna redovisas på Vården i siffror vartefter data finns tillgängligt. Data redovisas könsuppdelat och totalt, och för både region- och enhetsnivå när det är möjligt och relevant.
Relaterad information
Kompletterande underlag
Referenser
- Wijsenbeek M, Cottin V. Spectrum of Fibrotic Lung Diseases. N Engl J Med. 2020;383(10):958-68.
- Wijsenbeek M, Suzuki A, Maher TM. Interstitial lung diseases. Lancet. 2022;400(10354):769-86.
- Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022;205(9):e18-e47.
- Spagnolo P, Ryerson CJ, Putman R, Oldham J, Salisbury M, Sverzellati N, et al. Early diagnosis of fibrotic interstitial lung disease: challenges and opportunities. Lancet Respir Med. 2021;9(9):1065-76.
- Joy GM, Arbiv OA, Wong CK, Lok SD, Adderley NA, Dobosz KM, et al. Prevalence, imaging patterns and risk factors of interstitial lung disease in connective tissue disease: a systematic review and meta-analysis. Eur Respir Rev. 2023;32(167).
- Graney BA, Fischer A. Interstitial Pneumonia with Autoimmune Features. Ann Am Thorac Soc. 2019;16(5):525-33.
- Rajan SK, Cottin V, Dhar R, Danoff S, Flaherty KR, Brown KK, et al. Progressive pulmonary fibrosis: an expert group consensus statement. Eur Respir J. 2023;61(3).
- Sköld M (red). Idiopatisk lungfibros. Vårdprogram. Göteborg: Mediahuset/Svensk lungmedicinsk förening; 2019.
- Kreuter M, Picker N, Schwarzkopf L, Baumann S, Cerani A, Postema R, et al. Epidemiology, healthcare utilization, and related costs among patients with IPF: results from a German claims database analysis. Respir Res. 2022;23(1):62.
- Podolanczuk AJ, Thomson CC, Remy-Jardin M, Richeldi L, Martinez FJ, Kolb M, et al. Idiopathic pulmonary fibrosis: state of the art for 2023. Eur Respir J. 2023;61(4).
- Ferrara G, Arnheim-Dahlström L, Bartley K, Janson C, Kirchgässler K-U, Levine A, et al. Epidemiology of Pulmonary Fibrosis: A Cohort Study Using Healthcare Data in Sweden. 2019;5(1):55-68.
- Perelas A, Silver RM, Arrossi AV, Highland KB. Systemic sclerosis-associated interstitial lung disease. Lancet Respir Med. 2020;8(3):304-20.
- Wallace B, Vummidi D, Khanna D. Management of connective tissue diseases associated interstitial lung disease: a review of the published literature. Curr Opin Rheumatol. 2016;28(3):236-45.
- Lescoat A, Huscher D, Schoof N, Airò P, de Vries-Bouwstra J, Riemekasten G, et al. Systemic sclerosis-associated interstitial lung disease in the EUSTAR database: analysis by region. Rheumatology (Oxford). 2023;62(6):2178-88.
- Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18-64 years old. Eur Respir J. 2016;48(1):179-86.
- Distler O, Assassi S, Cottin V, Cutolo M, Danoff SK, Denton CP, et al. Predictors of progression in systemic sclerosis patients with interstitial lung disease. Eur Respir J. 2020;55(5).
- Meyer KC. Pulmonary fibrosis, part I: epidemiology, pathogenesis, and diagnosis. Expert Rev Respir Med. 2017;11(5):343-59.
- Borie R, Kannengiesser C, Antoniou K, Bonella F, Crestani B, Fabre A, et al. European Respiratory Society statement on familial pulmonary fibrosis. Eur Respir J. 2023;61(3).
- Cottin V, Valenzuela C. Diagnostic approach of fibrosing interstitial lung diseases of unknown origin. Presse Med. 2020;49(2):104021.
- Dowman L, Hill CJ, May A, Holland AE. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst Rev. 2021;2(2):Cd006322.
- Overgaard D, Kaldan G, Marsaa K, Nielsen TL, Shaker SB, Egerod I. The lived experience with idiopathic pulmonary fibrosis: a qualitative study. The European respiratory journal. 2016;47(5):1472.
- Nolan CM, Polgar O, Schofield SJ, Patel S, Barker RE, Walsh JA, et al. Pulmonary Rehabilitation in Idiopathic Pulmonary Fibrosis and COPD: A Propensity-Matched Real-World Study. Chest. 2022;161(3):728-37.
- Lindell KO, Klein SJ, Veatch MS, Gibson KF, Kass DJ, Nouraie M, et al. Nurse-Led Palliative Care Clinical Trial Improves Knowledge and Preparedness in Caregivers of Patients with Idiopathic Pulmonary Fibrosis. Ann Am Thorac Soc. 2021;18(11):1811-21.
- Kataoka K, Nishiyama O, Ogura T, Mori Y, Kozu R, Arizono S, et al. Long-term effect of pulmonary rehabilitation in idiopathic pulmonary fibrosis: a randomised controlled trial. Thorax. 2023;78(8):784-91.
- Nakazawa A, Dowman LM, Cox NS, Brazzale DJ, McDonald CF, Hill CJ, et al. Prescribing walking training in interstitial lung disease from the 6-minute walk test. Physiother Theory Pract. 2023;39(4):873-7.
- Holland AE, Spruit MA, Troosters T, Puhan MA, Pepin V, Saey D, et al. An official European Respiratory Society/American Thoracic Society technical standard: field walking tests in chronic respiratory disease. Eur Respir J. 2014;44(6):1428-46.
- Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am J Respir Crit Care Med. 2016;194(3):265-75.
- Oishi K, Matsunaga K, Asami-Noyama M, Yamamoto T, Hisamoto Y, Fujii T, et al. The 1-minute sit-to-stand test to detect desaturation during 6-minute walk test in interstitial lung disease. NPJ Prim Care Respir Med. 2022;32(1):5.
- Enright, P. L. & Sherrill, D. L. Reference equations for the six-minute walk in healthy adults. Am J Respir Crit Care Med 158, 1384-1387 (1998).
- Strassmann, A. et al. Population-based reference values for the 1-min sit-to-stand test. Int J Public Health 58, 949-953 (2013).
- Wapenaar M, Patel AS, Birring SS, Domburg RTV, Bakker EW, Vindigni V, et al. Translation and validation of the King’s Brief Interstitial Lung Disease (K-BILD) questionnaire in French, Italian, Swedish, and Dutch. Chron Respir Dis. 2017;14(2):140-50.
- Norrby E, Lindahl I. Dialog om arbetsförmåga (DOA), version 5.0 inklusive DOA-lättläst. Sveriges arbetsterapeuter. 2023.
- McColl MA, Denis CB, Douglas KL, Gilmour J, Haveman N, Petersen M, et al. A Clinically Significant Difference on the COPM: A Review. Can J Occup Ther. 2023;90(1):92-102.
- Tornquist K, Sonn U. ADL-Taxonomin® - en bedömning av aktivitetsförmåga. Sveriges arbetsterapeuter. 2022.
- Borie R, Kannengiesser C, Antoniou K, Bonella F, Crestani B, Fabre A et.al. European Respiratory Society statement on familial pulmonary fibrosis. Eur Respir J. 2023 Mar 16;61(3):2201383. Doi: 10.1183/13993003.01383-2022. PMID: 36549714
- Hunninghake GM, Goldin JG, Kadoch MA, Kropski JA, Rosas IO, Wells AU, et al.; ILA Study Group. Detection and Early Referral of Patients With Interstitial Lung Abnormalities: An Expert Survey Initiative. Chest. 2022;161(2):470-482.
Konsekvensbeskrivning
Bilagor
Bilaga 1– Kompletterande bakgrund om interstitiell lungsjukdom (ILD). VF Lungfibros.pdf
Bilaga 2 – Autoimmun sjukdom . VF Lungfibros. docx.pdf
Bilaga 3 – Interstitiella lungabnormaliteter (ILA) VF Lungfibros. docx.pdf
Bilaga 4 – Anamnes. VF Lungfibros. docx.pdf
Bilaga 5 – Klinisk undersökning och utredning. VF Lungfibros. docx.pdf